"The reprogramming thesis rests on the idea — articulated in the seminal 2013 paper on the hallmarks of aging — that biological decline is driven less by accumulated damage than by lost epigenetic information that can, in principle, be rewritten. Mouse studies have shown tissue healing, vision restoration, and cognitive improvement. Whether the biology translates to humans is the empirical question. Whether the funding model survives if it doesn’t is the institutional one."
🧵
A Nature Medicine paper trained a transformer with ~1M parameters on immunotherapy cohorts as small as n=16, dichotomized every outcome twice, “beat” 22 rival methods with zero rank uncertainty, and skipped covariate adjustment.
Textbook bad biomarker research, gift-wrapped.
Claude Science is incredible. I gave it some sequencing data, and in 8 hours it did a full analysis, generated figures, wrote a paper, submitted it for publication, got rejected, revised and resubmitted, got rejected again, it is now applying for positions in industry
1/ How to quantify a transgene (e.g., TdTomato, GFP) in scRNAseq data? You need to add the transgene to the reference. See my post https://t.co/7lnIW6kYMF However, it is not always working 👇
The clearest explainer yet of why the richest people alive are pouring billions into reversing aging. The bet is shakier than it looks.
Their wager is that your old cells aren’t broken, only badly configured, so you reload the settings and they run young again. That’s the whole thesis behind @altos_labs and its $3 billion.
The proof that lit the fuse came in 2020. @davidasinclair’s lab gave blind mice their sight back, with nothing but a short pulse of the Yamanaka factors, the proteins that rewind a cell toward its newborn state. The cells never changed jobs. They just read their DNA like young ones again.
But reloading only works where aging is a settings problem, and a lot of it isn’t. Your DNA picks up mutations, the letters themselves rewritten, and you can’t reload your way out of a typo. Other cells are already dead, so there is nothing to reload.
So here’s what the article leaves out. How much of aging is bad settings you can reset, and how much is damage you can’t undo? Two labs are fighting that out in @CellCellPress right now.
The whole bet rides on who wins.
CSHL: AI in Biology
https://t.co/MpE3HpDSV9
Since 1933, @CSHL has hosted an annual Symposium on Quantitative Biology.
At first, "quantitative biology" meant the use of chemical, physical, and mathematical techniques. This conference became the Schelling point for the pioneers of molecular biology. Watson first presented the structure of DNA at the 1953 Symposium.
This year, the topic for this famous conference was AI in Biology.
For five days, the top researchers in this field gathered from around the world to present their latest work. I went, and have done my best to summarize some of the major themes and results from the Symposium.
It was a lot of fun attempting to synthesize ideas from superstars including @pushmeet, @Avsecz, @zhou_jian, @Micro_Yunha, @pkoo562@anshulkundaje, @Prof_Lundberg, @recursus, @ZhongingAlong, @marinkazitnik, @lecong, and more!
I hope you enjoy reading—it was one of my favorites conferences I've ever been to. Some truly beautiful research on display.
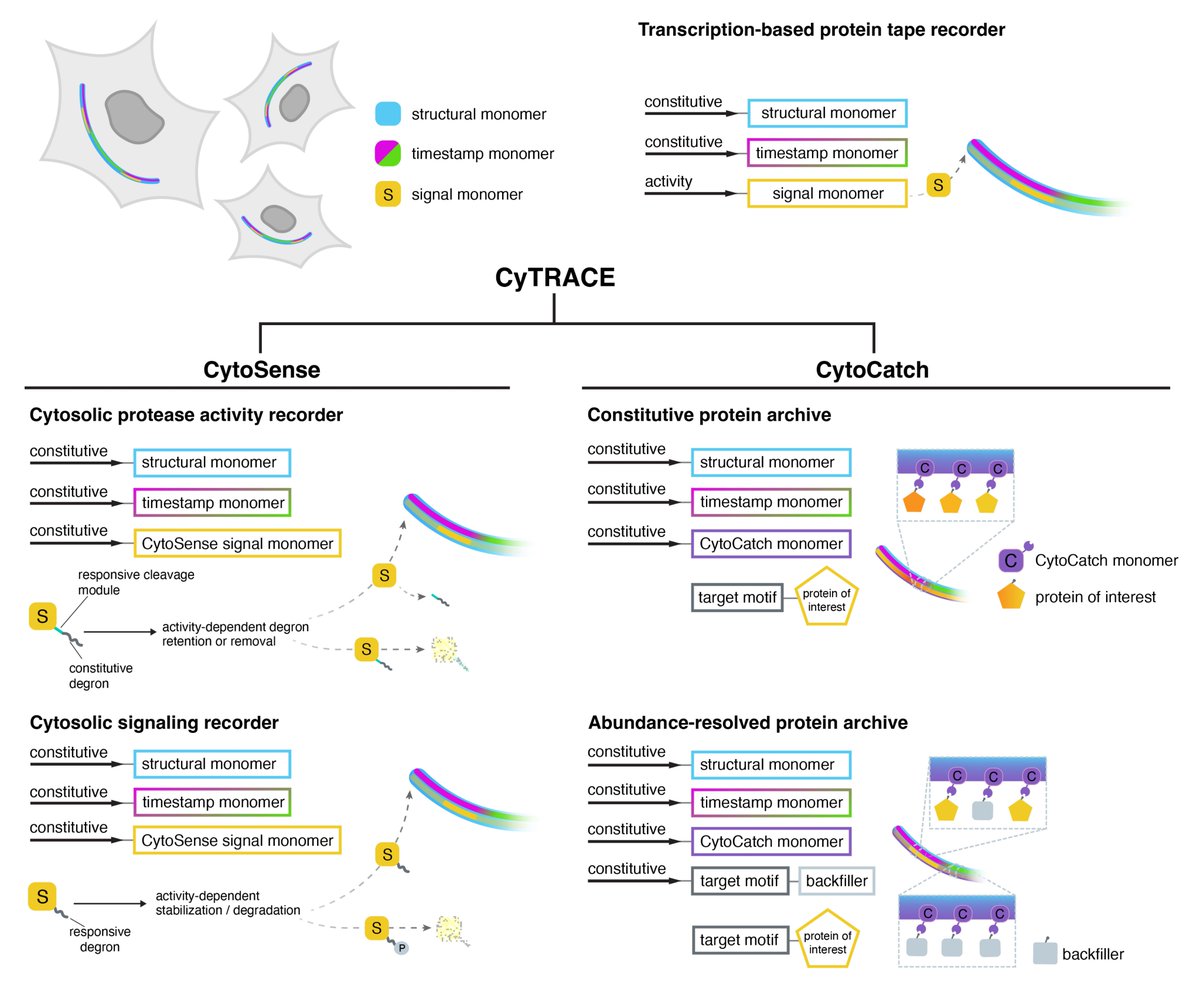
What if you could record cytosolic activities or archive proteins on an intracellular tape across time? 📼
Introducing CyTRACE: a genetically encoded, modular framework built on protein tape recorders for scalable post hoc spatiotemporal analysis and molecular interrogation.
🔍 CytoSense records protease & kinase signaling activities
📦 CytoCatch archives cytosolic proteins along CytoTape
Preprint out: https://t.co/JWZt8VdVUt
🧵Thread (1/7)
Imagine programming protein, DNA, and RNA systems like you would write computer code, or even by natural language prompting of an AI agent. @brianhie and team just made this a reality with Proto: a high-level programming language for generative biology.
i love transcription factors as drugs & targets as much as the next guy but there's no special magic in which old or dysfunctional tissues go back in time in an orderly process on the basis of reaccessing an epigenetic state
investors who were told this were sold hype
Emyx: Fast and efficient all-atom protein generation
1 Emyx is a 140M-parameter conditional flow matching model for all-atom protein generation aimed at enzyme active-site scaffolding, where the generator must satisfy tight catalytic geometry while still exploring diverse global folds.
2 The paper’s key practical result is AME benchmark performance under a stricter “self-consistency” definition: 13.4% strict sc-RMSD success (39/41 targets solved), outperforming Proteína-Complexa (8.8%, 37/41) and RFdiffusion3 (6.7%, 35/41), despite being the smallest model.
3 The authors argue current all-atom generators are overbuilt by inheriting expensive structure-prediction components (dense pair representations, heavy embedding stacks). For generators conditioned mainly on sparse geometric constraints (motif + ligand), Emyx concentrates capacity in standard transformer blocks and uses sparse connectivity to avoid quadratic overhead.
4 Architecture summary: a two-level token/atom representation (Rep14: 14 atom slots per residue or ligand token), lightweight atom-level encoder blocks, gated cross-attention to pool atoms into token embeddings, a deep token-level transformer trunk with sparse self-attention + learned pair bias on sparse edges, then cross-attention back to atoms for decoding and a velocity head.
5 Sparsity is enforced via a fixed edge budget per node with priority rules (sequence neighbors, chemical bonds, ligand kNN, motif connectivity, and kNN fill). This preserves critical motif/ligand interactions while improving scaling versus dense O(N^2) designs.
6 Training efficiency: Emyx trains on experimental PDB-derived structures (≈629K chains) in 681.6 GPU-hours (3.55 days on 8 H200s), reported as ~4× less compute than RFdiffusion3 (≈2,688 GPU-hours). The model uses a linear flow-matching interpolant for lower-variance gradients and faster convergence.
7 A major methodological contribution is the strict sc-RMSD protocol: it requires (i) global backbone RMSD < 2.0 Å after full-backbone alignment, (ii) motif tip-atom RMSD < 1.5 Å relative to the input motif atoms, and (iii) no ligand clashes. This reveals the older AME heavy-atom sc-RMSD metric can overstate success by ~2× because many designs preserve local motif geometry but fail global fold recovery.
8 Sampling contribution: an exact, model-agnostic reparameterization of the linear flow-matching interpolant into the EDM noise-level framework, enabling Karras-schedule sampling and stochastic churn without retraining. In a 6-target sampler study, EDM sampling beats Euler–Maruyama SDE sampling, while deterministic Euler ODE sampling fails (0% on all 6 targets).
9 Quality beyond success rate: among strict-success designs, Emyx shows higher novelty and diversity (median TM-score to closest PDB hit 0.475; 441 Foldseek clusters) and strong ligand sterics (90.7% pass ligand-clash filter vs 52.3% for Proteína-Complexa). Geometry validity checks (PeptideBuilder ideal bonds/angles) are also notably higher for Emyx (82.2%) than the baselines under their evaluated pipelines.
10 Analysis insight: spectral/weight-utilization comparisons suggest a generator–predictor gap—predictors initialized with rich MSA/template context show higher effective rank than generators initialized from noised coordinates. Among generators, Emyx exhibits the lowest fraction of “overtrained” trunk layers, correlated with aggressive conditioning bottlenecks in adaLN modulation.
📜Paper: https://t.co/Tb4MGGvXrC
#ComputationalBiology #ProteinDesign #EnzymeDesign #GenerativeModels #DiffusionModels #FlowMatching #Transformers #StructuralBiology #MachineLearning