Identification of Pathogenic Copy Number Variants in Mexican Patients With Inherited Retinal Dystrophies Applying an Exome Sequencing Data‐Based Read‐Depth Approach - Fabian‐Morales - 2024 - Molecular Genetics & Genomic Medicine - Wiley Online Library https://t.co/6BkiaUV37k
What is a reference genome? 🧬
In the context of HPRC, a reference genome is a low-error genome and associated annotation coordinate system that serves as the backbone for genome alignment.
Explore more HPRC definitions: https://t.co/YneaCJZy3n
Mwenda Rintari from Exeter presents his data on spliceogenic missense. 2.7% of missense variants in UK Biobank had a spliceAI > 0.2 and 72% of these also have low missense pathogenicity predictions with REVEL < 0.5 and AlphaMissense < 0.34. These variants did have an effect of protein levels suggesting NMD (when correcting for misbinding of antibodies to misfolded proteins w/o NMD).
So yeah, check splicing score on your missense variants. #eshg2026
🚨 Breaking neuroscience/evolution news!
New Nature paper provides further evidence for a central hypothesis (with a long lineage) that I defended in my 2004 book, The Birth of the Mind: the evolutionary process of duplication and divergence played a key role in brain evolution.
I watched the recording of the recent @gnomad_project@GA4GH webinar on v4 to address common misconceptions about the gnomAD by @HeidiRehm@SamBaxterCGC and Katherine Chao. 👀 📺
Much of that was said is likely known to most who use gnomAD on a daily basis such as myself. Still, some interesting nuggets were in there. So, here are my unsorted notes, in case you find them helpful:
gnomAD is not a control group in the normal sense, but represents a reasonable reference set for severe diseases such as child-onset muscular dystrophy. It is best thought of as a general population dataset. It is NOT a healthy population dataset.
Included are individuals from case-control studies and biobank participants. Not included are cohorts specifically recruited for detection of early onset mendelian diseases to avoid any over-enrichment of causal variants.
Under US regulatory framework, there is no chance of any HPO terms that can be made available by a resource such as gnomAD. Because its freely accessible. No login required. Only sex, age and original study that the person was recruited in, is available to the gnomAD team. Only aggregated data can be shared. Not details on any specific participant.
The only description they have is, if a person is a "control" or "case" in the original study and that only refers to the small phenotypic window thats addressed in the original study (say schizophrenia or diabetes).
This is in contrast to @uk_biobank or @AllofUsResearch data. These datasets are not freely accessible - you only get to access them through their data portal where you need to login. But you get individual level data.
@HeidiRehm said it will be "a while" until gnomAD data is available on another genome build other than the current hg38. Conversations on e.g. T2T or pangenome graph type of future gnomAD versions have just begun.
Roughly half of the exome data made available in gnomAD v4 is from @uk_biobank. As it is a biobank that has a healthy volunteer bias, because volunteers tend to be slightly wealthier and healthier than the general population (see PMID: 28641372). Which actually means that the non-UK-biobank subset in gnomAD has a higher disease burden because it is more broadly recruited.
UK Biobank participants do have rare diseases, see PMID: 28641372
Individuals in gnomAD are also from case-control studies, such as schizophrenia, myocardial infarction etc. Thus the gnomAD team created subsets, such as non-neuro to filter out certain groups/study participants. See contributing projects here https://t.co/Rll5B28ZBt
At the current size of gnomAD in v4, any phenotype enrichment is less of a concern than in smaller previous versions. Simply because the dataset is 800k people.
Question - should a subset be used to apply e.g. the ACMG BA1 or BS1 criteria? - @HeidiRehm says v4 is best, when used as a whole dataset, because the more people, the better. If you want a max allele freq for a certain variant, then check ancestry subgroups.
All of gnomAD v4 can be considered as roughly "non-cancer" because all of the data from TCGA (The Cancer Genome Atlas Program) was removed due to QC. Which means, people in gnomAD can of course have cancer, but there is no enrichment of cancer phenotypes in v4.
Question - there seems to be higher freq of cancer variants in the non-UK-biobank subset. Why? - KC: Could be due to recruiting, since UK Biobank only recruited people aged 40-69 years old. They did find far lower rates of cancer in UKB than in the general UK population. Again, biobank participants are on average healthier.
@AllofUsResearch data will be inclued in v5 (+ another 800k individuals), which is "coming soon". Again, no phenotype data.
Question - what about CHIP variants? - @HeidiRehm: gnomAD has a full spectrum of age so likely no enrichment, but of course older people are in there with CHIP variants. There are flags on genes, to signal high rate of CHIP variants.
gnomAD does have age on many individuals included, but not all. This was information given by the original study. It is unclear to the gnomAD team actually what is meant by age in all of the different studies - e.g. time of inclusion into the study? Age when data was transferred to gnomAD? Other? The gnomAD team did not tinker with the age data, just used it as is, where possible and shares it in buckets in individual variant pages.
Question - are you planning to include the million variant program dataset? - @HeidiRehm - not yet included because of no easy access to it.
Since external sequencing data has to be reprocessed by the gnomAD team (quite expensive), only highly "valuable" datasets, meaning ancestrally diverse external datasets will be re-processed in future gnomAD. Simply due to cost-benefit.
gnomAD v4 is mostly saturated for common variants from european ancestries, so non-european ancestries are more "valuable". Here is the preprint for gnomAD v4 by @konradjk and team if you want to dive into that PMID: 41929314.
Question - why is #CFTR delta-F508 58x homozygous in gnomAD? @HeidiRehm - the team did a little research, as this is likely a bit too high for a general population. Turns out, a cohort from a clinic (important: not from UK Biobank) was included with a pulmonary focus.
https://t.co/v89wQnylOi
My former colleague @tomaeusTo also stumbled upon this #CFTR variant back in 2023 right after the release. See here for more details with a link to the gnomAD FAQ forum:
https://t.co/YJIZ1bOUVw
There are actually more than a few #CFTR over-represented variants in gnomAD, again likely due to the inclusion of that one pulmo clinic's cohort.
Question - what about LongReads? HR: @TalkowskiLab is working on a LongRead gnomAD dataset. It will happen. They are working on it. No date for release just yet.
There is a Canadian gnomAD dataset, not yet included in the primary gnomAD dataset. For now, it is available for download — with a browser interface coming soon. They actually want to provide phenotypes, but that is still "quite a few months out" (Jordan Lerner-Ellis).
https://t.co/ScPFH5Ug4b
https://t.co/M808TqEh3Q
https://t.co/SF8iN3NcKV
There is also a Singapore gnomAD dataset - probably not possible to access the data right now. So different groups can add their own wrinkles to these separate gnomad type datasets.
Recruiting more samples for gnomAD is ongoing. Federated gnomAD explained here: https://t.co/Xa9vLjU75e
@HeidiRehm points out there is also a gnomAD forum / discussion board. Please ask questions or feature requests. Link: https://t.co/bNUU98wOZ4
Thanks to the gnomAD on all their work on this invaluable resource. Couldn't do without it.
Hope this summary helps. 🙂
How can we help scientists get better data out of their instruments?
Using @GoogleDeepMind AlphaEvolve, @GoogleResearch identified new ways to train DeepConsensus, improving DNA sequencing accuracy and output on PacBio instruments.
Read more: https://t.co/BadpMAUmAu
Despite widespread clinical testing, many patients with rare genetic diseases do not receive a definitive molecular diagnosis.
Researchers report a new strategy using long-read RNA sequencing that improves the genetic diagnosis of rare diseases. https://t.co/f3GW0iE1af
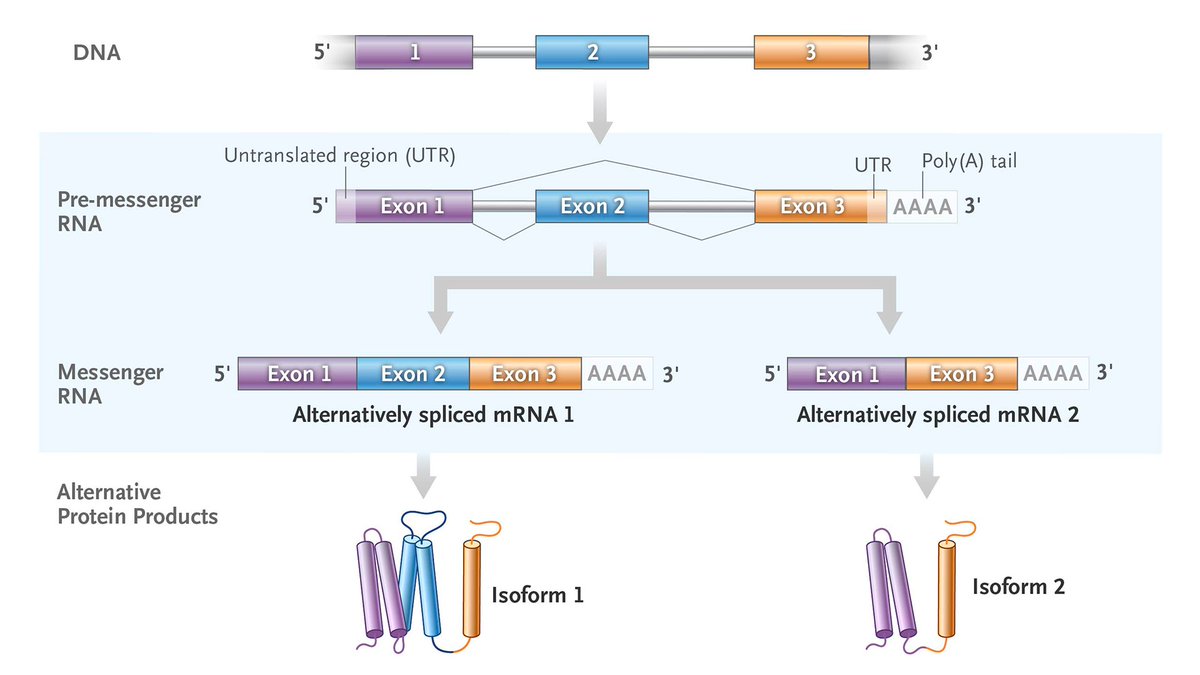
𝐀𝐥𝐭𝐞𝐫𝐧𝐚𝐭𝐢𝐯𝐞 𝐬𝐩𝐥𝐢𝐜𝐢𝐧𝐠 is the use of different exons in the formation of messenger RNA from initially identical transcripts, which can result in the generation of related proteins from one gene, often in a manner specific to a type of tissue or a developmental stage.
To learn more about this NEJM Illustrated Glossary term, read the editorial “Toward a Disease-Modifying Therapy for Dravet Syndrome” by Gemma L. Carvill, PhD, and Heather C. Mefford, MD, PhD, from @NUFeinbergMed and @StJudeResearch: https://t.co/78EEAj0GY8
Explore more terms: https://t.co/9j3iLbbGVo
Second time the MX biobank project (https://t.co/qaLnfPzcz2) gets the cover in a @Nature journal! This time in @NatureMedicine highlighting the work by @BarjonCar on Clinical genetic variation in Mexico. Another amazing cover by @mauguz33! https://t.co/k3eGLX4809
Veo a menudo que hay una confusión muy grande entre lo que es un embedding y un token.
Un token es simplemente una unidad de texto.
Puede ser una palabra, una sub-palabra o incluso un carácter, dependiendo del modelo. Es la forma en la que el texto se fragmenta para poder ser procesado.
Ejemplo:
“inteligencia artificial” → ["intel", "igencia", " artificial"]
El token no “entiende” nada por sí mismo. Solo es una pieza de texto.
Un embedding, en cambio, es una representación numérica de ese token (o de un conjunto de tokens) en un espacio vectorial.
Es ahí donde aparece el significado: tokens con contextos o usos similares terminan cerca entre sí en ese espacio.
Dicho de forma simple:
•Token = qué entra al modelo
•Embedding = cómo el modelo lo representa internamente
Confundirlos es como confundir una letra con el significado de una palabra completa.
Uno es la materia prima; el otro es la geometría que permite razonar, comparar y generalizar.
Si quieres entender cómo funcionan realmente los modelos de lenguaje, esta distinción es básica. Sin embeddings, los tokens son solo texto partido. Sin tokens, no hay nada que representar.
Introducing Prism, a free workspace for scientists to write and collaborate on research, powered by GPT-5.2.
Available today to anyone with a ChatGPT personal account: https://t.co/9mTLAbxPdH
PAREN TODO. Estás ante "el hallazgo de la década" en México. 🇲🇽😱
En Oaxaca, acaba de emerger una tumba zapoteca del año 600 d.C. que ha dejado a los expertos en shock por su conservación.
Guarda este hilo de datos para dimensionar esta joya de nuestra historia: 👇
📜 El Guardián: La figura central no es humana. Es un BÚHO (símbolo de la noche y el inframundo) cuyo pico "devora" o protege el rostro de un antiguo señor zapoteca.
🎨 Color Intacto: Después de 1,400 años, aún se conservan murales con pigmentos originales azules, verdes y ocres que narran procesiones rituales.
📍 No es Monte Albán: El hallazgo es en San Pablo Huitzo. Esto reescribe la historia, demostrando la complejidad y riqueza de los Valles Centrales fuera de la gran capital.
Una muestra brutal de la grandeza milenaria de la cultura Zapoteca. Piel de gallina.
📸: @INAHmx
El servicio de soporte solo se limitó a decir que el viaje no es elegible para un ajuste. Espero se pueda arreglar esta situación. El chofer es Miguel Ángel y el viaje temrina en Iztapalapa, un lugar totalmente diferente a mi destino. Espero me puedan dar una mejor atención.
Cobro de un viaje que nunca hice: El día de ayer (14 de enero) solicité un viaje por la noche, sin embargo el chofer nunca pasó por mi y tampoco canceló el viaje. El día de hoy me llegó el cargo $2863. El viaje quedo activo por horas @Uber_MEX@Uber_Support@Profeco















































![leonpalafox's tweet photo. Veo a menudo que hay una confusión muy grande entre lo que es un embedding y un token.
Un token es simplemente una unidad de texto.
Puede ser una palabra, una sub-palabra o incluso un carácter, dependiendo del modelo. Es la forma en la que el texto se fragmenta para poder ser procesado.
Ejemplo:
“inteligencia artificial” → ["intel", "igencia", " artificial"]
El token no “entiende” nada por sí mismo. Solo es una pieza de texto.
Un embedding, en cambio, es una representación numérica de ese token (o de un conjunto de tokens) en un espacio vectorial.
Es ahí donde aparece el significado: tokens con contextos o usos similares terminan cerca entre sí en ese espacio.
Dicho de forma simple:
•Token = qué entra al modelo
•Embedding = cómo el modelo lo representa internamente
Confundirlos es como confundir una letra con el significado de una palabra completa.
Uno es la materia prima; el otro es la geometría que permite razonar, comparar y generalizar.
Si quieres entender cómo funcionan realmente los modelos de lenguaje, esta distinción es básica. Sin embeddings, los tokens son solo texto partido. Sin tokens, no hay nada que representar.](https://pbs.twimg.com/media/G_sxN81WAAA47Lr.jpg)























