1/ Thrilled to share a Perspective that summarizes 30 years of work from the many labs studying the human genetic and immunological causes of infectious diseases: https://t.co/jdgw3gOWl9
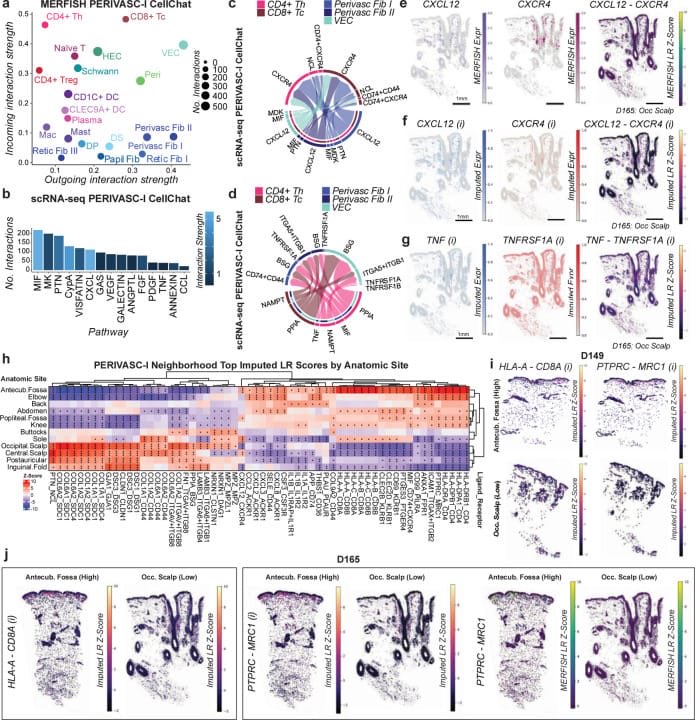
New @Nature Genetics paper mapped human skin at single-cell spatial resolution across 15 anatomic sites and 1.2 million cells — and the headline finding deserves attention:
Anatomy is encoded in cell states, not just cell types.
A fibroblast in your scalp and a fibroblast in your palm are both fibroblasts. Same cell type, same surface markers. But their gene expression programs are tuned to their location. The body doesn't just sort cells into the right places — it writes positional information directly into molecular state.
This has been hinted at in vitro for years (fibroblasts cultured from different body sites retain "positional memory"). What Ji lab did is put spatial context back in. Using MERFISH across 22 donors with matched sites, they could distinguish whether anatomic differences reflect cell abundance, cell state, or neighborhood architecture — and the answer is all three, operating together.
The cell-cell communication finding is equally striking. TNF emerges as a key regulator of immune-fibroblast crosstalk in a perivascular niche that resembles organized lymphoid tissue — right in the skin. When those neighborhoods break down, you get disease.
This points directly at one of the hardest unsolved problems in building single cells foundation models. Most single-cell models treat cells as isolated units — a transcriptomic snapshot divorced from where the cell lives and who its neighbors are. But biology doesn't work that way. A cell's state is partly a function of the signals it receives from adjacent cells, the extracellular matrix it sits in, and the tissue niche it belongs to. Spatial context and cell-cell interaction aren't annotations on top of the biology — they are the biology. Any virtual cell worth the name will need to internalize that. Atlases like this one are what that training data looks like.
Press release : https://t.co/Q0xNeRHD8y
Paper: https://t.co/ItEGlJmIGV
Check out our new paper! 🧐
A high-coverage Neandertal genome confirms small, isolated populations, Denisovan admixture, and extreme population structure accelerating differentiation, with levels matching the most divergent humans today.
https://t.co/UpugSLw7rq
Latest thoughts. Today in @JExpMed. We are disomic individuals genetically. Transcriptionally we need not be. Here I discuss what we know about this and propose Theory of Personal Evolution. @KidsAtColumbia@ColumbiaMed https://t.co/zTLo7lZ6gB
1/ We are thrilled to announce that our American branch of the Laboratory of Human Genetics of Infectious Diseases (HGID) will relocate to @UTSWMedCenter in Dallas, Texas, effective July 1, 2026 J
Excited to share our new paper in @NatureChemistry!
Building on our Rare Codon Recoding (RCR) work @ScienceMagazine, we developed a multi-rare-codon strategy enabling the simultaneous incorporation of up to 5 distinct ncAAs in mammalian cells!
https://t.co/tLp3wZTQ9U
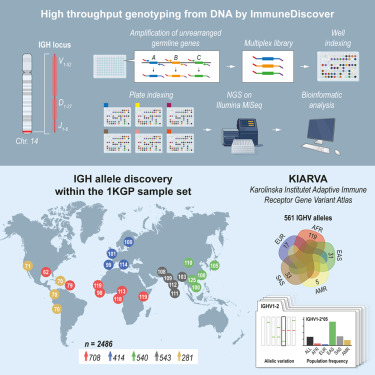
Ultra-high-throughput IGH genotyping of 25 global populations reveals population-biased allelic diversity and homozygous V and D gene deletions @ImmunityCP
https://t.co/evPNaYuA6c @NillaKH@karolinskainst
At @GoogleDeepMind, we believe AI is the ultimate catalyst for science. 🧬
The best example of this has been the AlphaFold database (AFDB) of protein structure predictions which has been used free of cost by more than 3.3 millions researchers across the world!
Today, in collaboration with @emblebi, @Nvidia and @SeoulNatlUni, we are expanding the database by adding millions of AI-predicted protein complex structures to the AlphaFold Database. To maximise global health impact, we’ve prioritised proteins that are important for understanding human health and disease, including homodimers from 20 of the most studied organisms, including humans, as well as the @WHO’S bacterial priority pathogens list.
Read more here:
https://t.co/RZGq8vrIPS
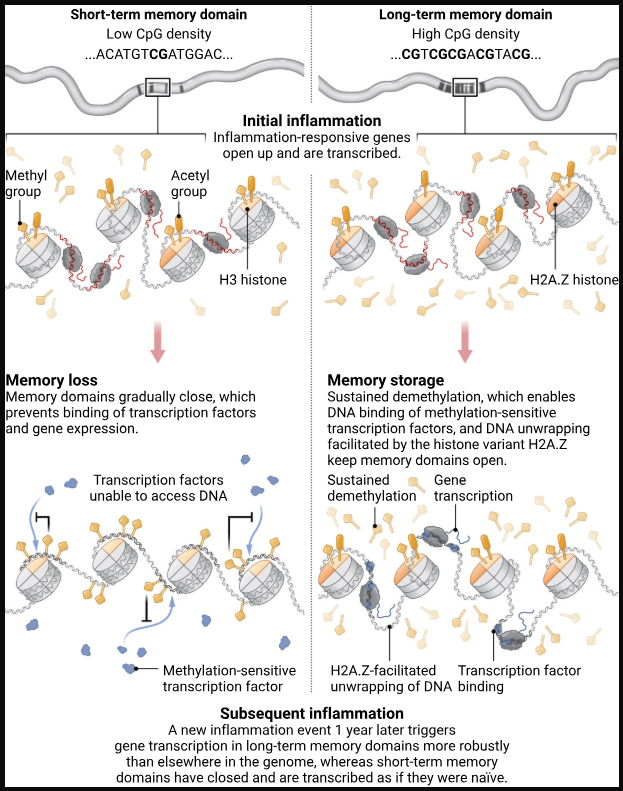
We are excited to announce the release of the Human Methylation Atlas Summary and Signals tracks for hg38 and hg19. The tracks display genome-wide DNA methylation profiles across 39 primary human cell types from 205 healthy tissue samples.
Learn more at https://t.co/GB3hxaYo1H
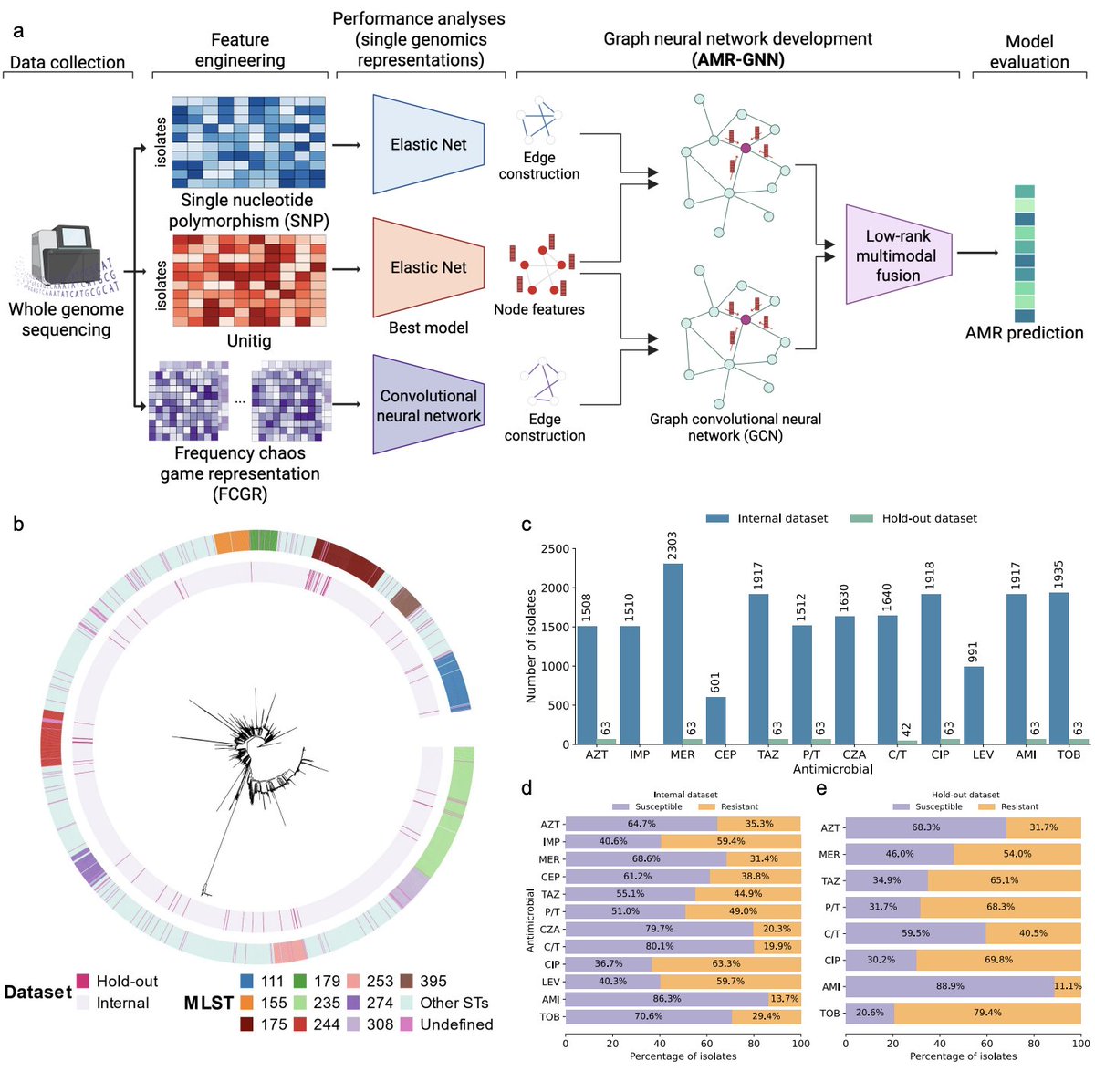
Graph neural networks that read bacterial genomes to predict antibiotic resistance
Antimicrobial resistance kills over a million people every year. When a patient arrives with a severe bacterial infection, clinicians need to know which antibiotics will work—fast. Culture-based susceptibility testing takes 2 to 5 days. Whole-genome sequencing offers a shortcut, but translating raw bacterial DNA into reliable resistance predictions is far from trivial.
Bacterial genomes can be represented in many ways—SNPs, reference-free unitigs, image-like frequency chaos game representations (FCGR)—and there is no consensus on which works best. Worse, bacteria reproduce clonally, so standard ML models often learn to recognise high-risk lineages rather than the actual resistance mechanisms.
Nguyen and coauthors tackle both problems with AMR-GNN, a graph neural network that integrates multiple genomic representations simultaneously. Unitig features serve as node features; SNP- and FCGR-derived pairwise distances define the graph edges. Two parallel GCN modules learn from the same nodes but different connectivity structures, and their embeddings are fused before a final resistance/susceptibility classification.
Tested on 2,515 Pseudomonas aeruginosa isolates across 12 antibiotics, AMR-GNN significantly outperforms single-representation models in 11/12 drugs—with AUROC gains of 28.8% for cefepime and 18.9% for aztreonam, precisely where prediction is hardest. A structural fix for clonal confounding—removing edges between isolates of the same sequence type, forcing the model to learn from genetically distinct neighbours—improves performance further across all tested antibiotics.
Validated on 23,000+ genomes spanning E. coli, K. pneumoniae, S. aureus, and E. faecium, mean AUROCs exceed 0.90 in nearly every species-drug combination. The model also recovers known resistance genes (gyrA, gyrB, parC for levofloxacin; fusA1 for tobramycin) through integrated gradient analysis—without any prior AMR knowledge encoded in the architecture.
Multi-representation learning, graph-based relational structure, and built-in interpretability. Three historically separate challenges, addressed in a single unified framework.
Paper: https://t.co/X4fa04j5CS
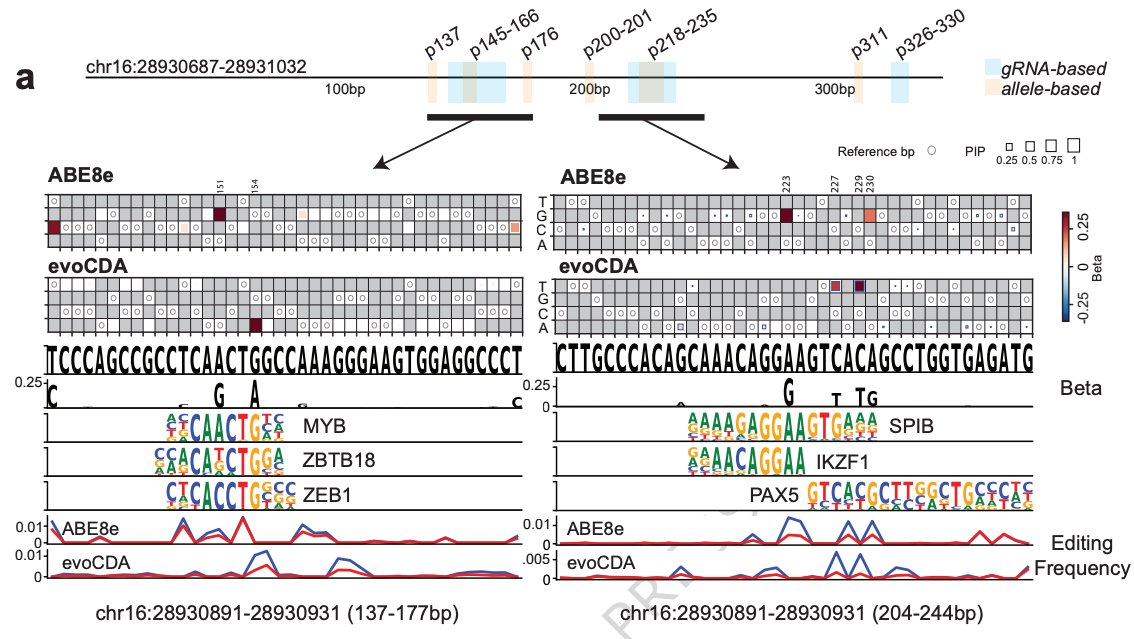
Excited to share another recent paper from the lab, in collaboration with @lucapinello and @danielevanbauer. We leveraged allele distributions after dense editing to identify the regulatory potential of individual nucleotides within #enhancers:
https://t.co/aTKlNX7P6U
Evo 2 is out in Nature today, showing that genome language models can predict and design across the full complexity of life, from phages to eukaryotes.
A few surprises from the project, including how ignoring trillions of nucleotides was key to getting a good model. 🧵
AlphaGenome is out in @nature today along with model weights! 🧬
📄 Paper: https://t.co/1fHzSPiY1x
💻 Weights: https://t.co/z6JWLT4Mpv
Getting here wasn’t a straight path. We sat down @googledeepmind to discuss the story behind the model, paper & API: https://t.co/cT8CiXfnxQ
🧬 A major data reanalysis in December's most-read Genetics paper uncovers thousands of previously hidden protein-coding regions in human and mouse genomes: https://t.co/CiRw4bRmPN