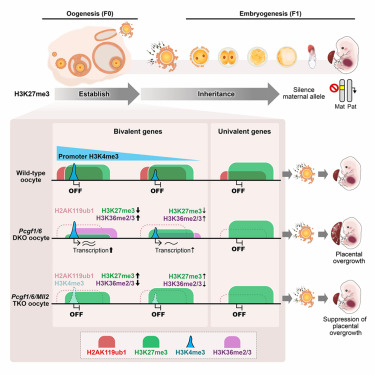
Today, we’re excited to share mRNAutilus, our experimentally-validated framework for multi-objective generation of full-length mRNAs, jointly optimizing coding sequences and UTRs for expression, stability, and translation!⚓️
📜: https://t.co/jld3bjXIOH
💻: https://t.co/z2K4l60THy
The paradigm says that coactivators are passively recruited to targets by transcription factors (TFs). But is that the whole story?
We explore the Mediator subunit Med15 to ask: Do its activator-binding domains (ABDs) alone drive UAS targeting?
https://t.co/A3yxJXj9zj
𝗧𝗼𝗱𝗮𝘆, 𝗦𝗰𝗿𝗶𝗯𝗲 𝗧𝗵𝗲𝗿𝗮𝗽𝗲𝘂𝘁𝗶𝗰𝘀 𝗼𝗳𝗳𝗶𝗰𝗶𝗮𝗹𝗹𝘆 𝗯𝗲𝗰𝗮𝗺𝗲 𝗮 𝗰𝗹𝗶𝗻𝗶𝗰𝗮𝗹-𝘀𝘁𝗮𝗴𝗲 𝗯𝗶𝗼𝘁𝗲𝗰𝗵𝗻𝗼𝗹𝗼𝗴𝘆 𝗰𝗼𝗺𝗽𝗮𝗻𝘆 𝘄𝗶𝘁𝗵 𝘁𝗵𝗲 𝗮𝗱𝘃𝗮𝗻𝗰𝗲𝗺𝗲𝗻𝘁 𝗼𝗳 𝗦𝗧𝗫-𝟭𝟭𝟱𝟬, a novel 𝘪𝘯 𝘷𝘪𝘷𝘰 epigenetic CRISPR therapy designed to deliver ultra-durable lowering of “bad cholesterol,” from a single dose, all without permanently altering the genome.
This program is built on years of intentional and iterative engineering focused on improving the safety, specificity, potency, and durability of CRISPR medicines.
𝗕𝘂𝘁 𝘄𝗵𝗮𝘁 𝗲𝘅𝗰𝗶𝘁𝗲𝘀 𝗺𝗲 𝗺𝗼𝘀𝘁 𝗶𝘀 𝘄𝗵𝗮𝘁 𝘁𝗵𝗶𝘀 𝗰𝗼𝘂𝗹𝗱 𝗺𝗲𝗮𝗻 𝗳𝗼𝗿 𝗽𝗮𝘁𝗶𝗲𝗻𝘁𝘀 𝗹𝗶𝗸𝗲 𝗺𝘆𝘀𝗲𝗹𝗳.
As someone at high risk of ASCVD, like roughly one-third of adults in the U.S., I’ve spent a lot of time thinking about the burden patients carry. For a chronic disease like ASCVD, prevention is far from easy. Success depends on maintaining near-perfect adherence to pills or injections for decades. In the real world, that’s incredibly difficult and simply not practical for most people. The fact that ASCVD remains the leading cause of death globally, despite plenty of therapeutic choices, makes that painfully clear.
𝗧𝗵𝗲 𝗳𝘂𝘁𝘂𝗿𝗲 𝗼𝗳 𝗺𝗲𝗱𝗶𝗰𝗶𝗻𝗲 𝘀𝗵𝗼𝘂𝗹𝗱 𝗮𝘀𝗽𝗶𝗿𝗲 𝘁𝗼 𝗺𝗼𝗿𝗲 𝘁𝗵𝗮𝗻 𝗰𝗵𝗮𝗶𝗻𝗶𝗻𝗴 𝗽𝗮𝘁𝗶𝗲𝗻𝘁𝘀 𝘁𝗼 𝗹𝗶𝗳𝗲𝗹𝗼𝗻𝗴 𝗺𝗲𝗱𝗶𝗰𝗮𝘁𝗶𝗼𝗻𝘀.
The vision behind STX-1150 is to provide year to decades of LDL-C lowering from a simple intervention, helping free patients from the constant burden of chronic treatment while more effectively reducing the risk of the world’s leading cause of death.
The future is about empowering patients to take greater control of our own health destiny and preventing disease rather than waiting to treat it after catastrophe occurs.
𝗜 𝗯𝗲𝗹𝗶𝗲𝘃𝗲 𝗮 𝗻𝗲𝘄 𝗲𝗿𝗮 𝗼𝗳 𝘁𝗵𝗲𝗿𝗮𝗽𝗲𝘂𝘁𝗶𝗰𝘀 𝗶𝘀 𝗼𝗻 𝘁𝗵𝗲 𝗵𝗼𝗿𝗶𝘇𝗼𝗻, 𝗼𝗻𝗲 𝘄𝗵𝗲𝗿𝗲 𝗺𝗲𝗱𝗶𝗰𝗶𝗻𝗲 𝗰𝗮𝗻 𝗱𝘂𝗿𝗮𝗯𝗹𝘆 𝗿𝗲𝘀𝗵𝗮𝗽𝗲 𝗹𝗼𝗻𝗴-𝘁𝗲𝗿𝗺 𝗵𝗲𝗮𝗹𝘁𝗵 𝗮𝗻𝗱 𝗮𝗹𝗹𝗼𝘄 𝘂𝘀 𝗮𝗹𝗹 𝘁𝗼 𝗹𝗶𝘃𝗲 𝗹𝗼𝗻𝗴𝗲𝗿, 𝗵𝗲𝗮𝗹𝘁𝗵𝗶𝗲𝗿 𝗹𝗶𝘃𝗲𝘀 𝘄𝗶𝘁𝗵 𝗴𝗿𝗲𝗮𝘁𝗲𝗿 𝗳𝗿𝗲𝗲𝗱𝗼𝗺.
Extremely proud of the entire Scribe team for advancing this vision. Excited for what comes next.
Five years ago we sketched out the designs for our first epigenome editors. Proud to see those sketches move into the clinic.
You can read more about the technology underpinning our epi-editors in this preprint we recently presented at ASGCT: https://t.co/SuSGvwgnPb
Announcing our first clinical trial.
Scribe has secured regulatory clearance from Australia’s @TGAgovau to initiate a first-in-human clinical study of STX-1150 for the treatment of hypercholesterolemia, a major driver of atherosclerotic cardiovascular disease (ASCVD).
Details🧵
I wrote this piece to promote thoughtful, respectful, and rational engagement with controversial science topics. I hope it fosters constructive dialogue in the scientific community—thank you for reading and sharing 🙏🏼 @NatRevImmunol
https://t.co/iSdbikwy9p
Protein/enzyme engineering has a severe bottleneck — and it's not in AI modeling or compute time.
It's in actually building and testing protein variants.
Proud to introduce our latest work, MIDAS: a way to go from primers to protein assays in mammalian cells in one day. 🧵
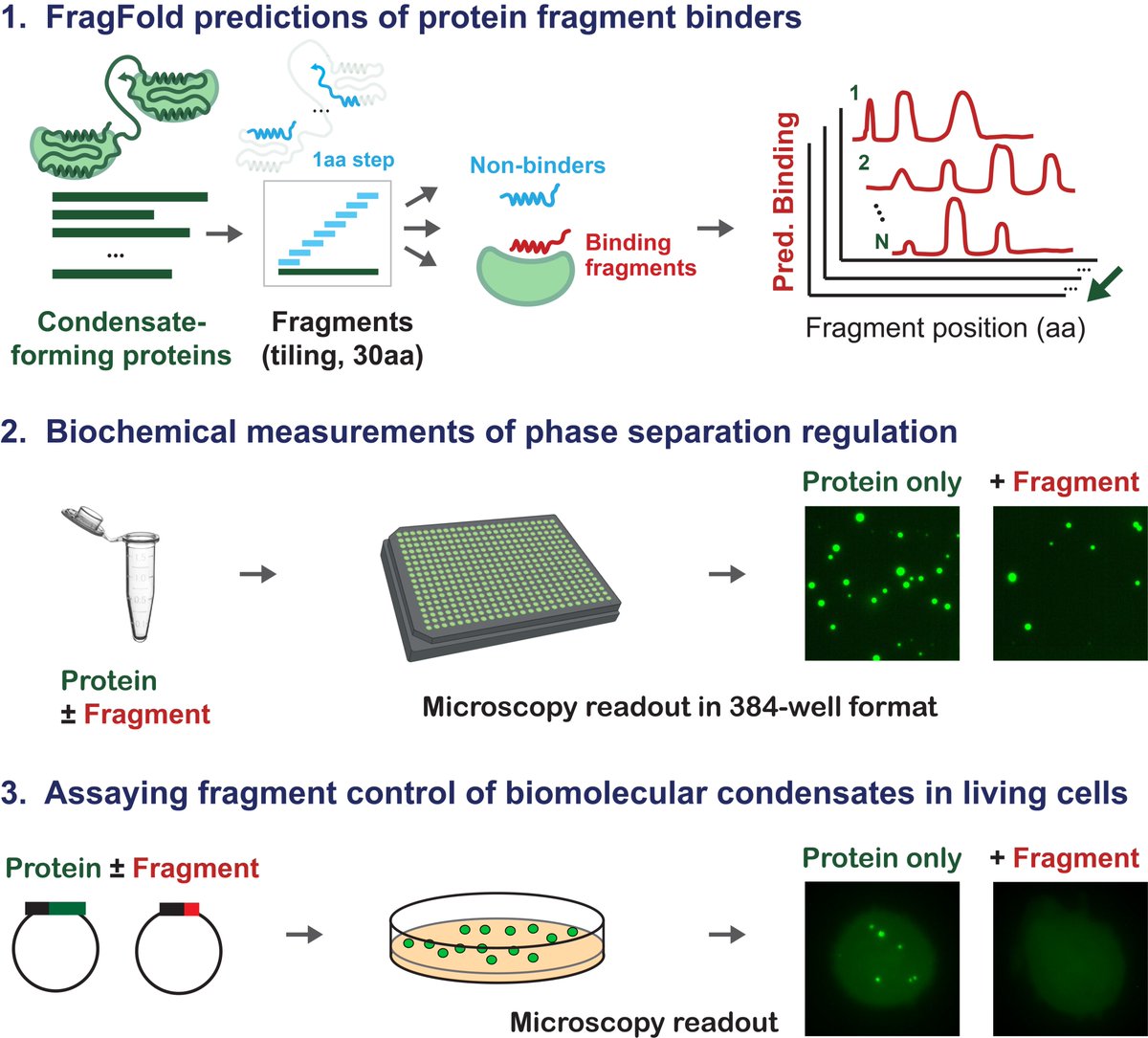
Interested in genetically encodable inhibitors of your favorite biomolecular condensate? Excited to announce our latest work, w/ @jibin_sadasivan, @GeneWeiLiLab, & @LindsayCase19, on protein fragments as generalizable regulators of phase separation. (1/n)
https://t.co/6IMkrTP3ZQ
"Human hepatocytes show continuous and lifelong turnover, allowing the liver to remain a young organ (average age <3 years)." Yet, "physiological liver cell renewal in humans is mainly dependent on diploid hepatocytes, whereas polyploid cells are compromised in their ability to divide." We get more polyploid cells as we age, and limit the ability of the liver to regenerate (at least so far): https://t.co/WYB55EhGXy.
"Given a fair wind [epigenetic editing] seems likely to establish itself as an important part of the medicine of the mid-21st century."
@TheEconomist recently ran a feature on epigenome editing, and the framing represents a major shift in mainstream understanding. They positioned the field not as a mere alternative to gene editing, but as the lower-risk, more nuanced path forward for many therapeutic applications.
Reflecting on this as someone who has been in the field since 2012, a few things stand out:
1. The "cutting paradigm" dominated the CRISPR conversation for years. The realization that epigenetic editing’s safer profile is a substantive scientific advantage is finally reaching a broader audience.
2. With three companies in human trials, including the progress we are making at Epicrispr Biotechnologies on FSHD, this is no longer speculative. We are in the era of patient outcomes.
3. The article rightfully flags longevity as a highly speculative frontier. While the epigenetic component of aging is real, we must remain ambitious without overpromising as we move from treating disease to enhancing human healthspan.
To the founders, clinicians, and especially the students and postdocs: this recognition is a signal that the work of the past decade has built something real.
The next decade will be even more transformative.
The article: https://t.co/ZSbv2Af53I
#EpigeneticEditing #Biotech #CRISPR #GeneTherapy #FSHD #EpicrisprBiotechnologies #Stanford
Our paper with Vijay Ramani is out today in @Nature.
We show that chromatin has a richer grammar than simple "open" or "closed" DNA. Using IDLI, we read 14 nucleosome structural states across single chromatin fibers and find that this grammar is actively written by transcription factors.
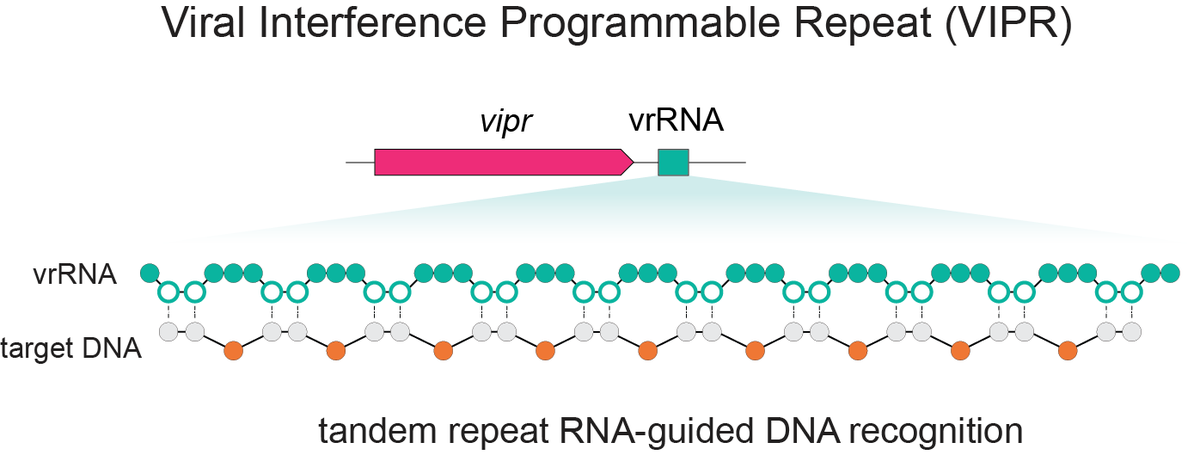
Excited to share our discovery of a new programmable RNA-guided DNA-targeting system hiding inside bacteriophages that predates CRISPR.
We call it VIPR (Viral Interference Programmable Repeat), and it uses an entirely new logic to find its targets.
Thread + link below.
How does a cell learn? In our new perspective in @Nature we propose a model where the properties of the AP-1 family of transcription factors – stress-induced feedback, regulatory combinatorics & cellular memory – encode a mechanism for cellular learning. https://t.co/Hzmq319EaD
Efficient genome editing with chimeric oligonucleotide-directed editing
"a DNA-dependent DNA polymerase paired with a chimeric pegRNA may address some of the limitations of current reverse transcriptase-based prime editors."
https://t.co/fma06wIPJG
1/n
🤩 Beyond excited to share our recent paper, published today @NatureComms!! 🤩
This study represents, in my view, one of the most striking phenotypes we ever found with really important findings and implications.
https://t.co/zG6QqPPEua
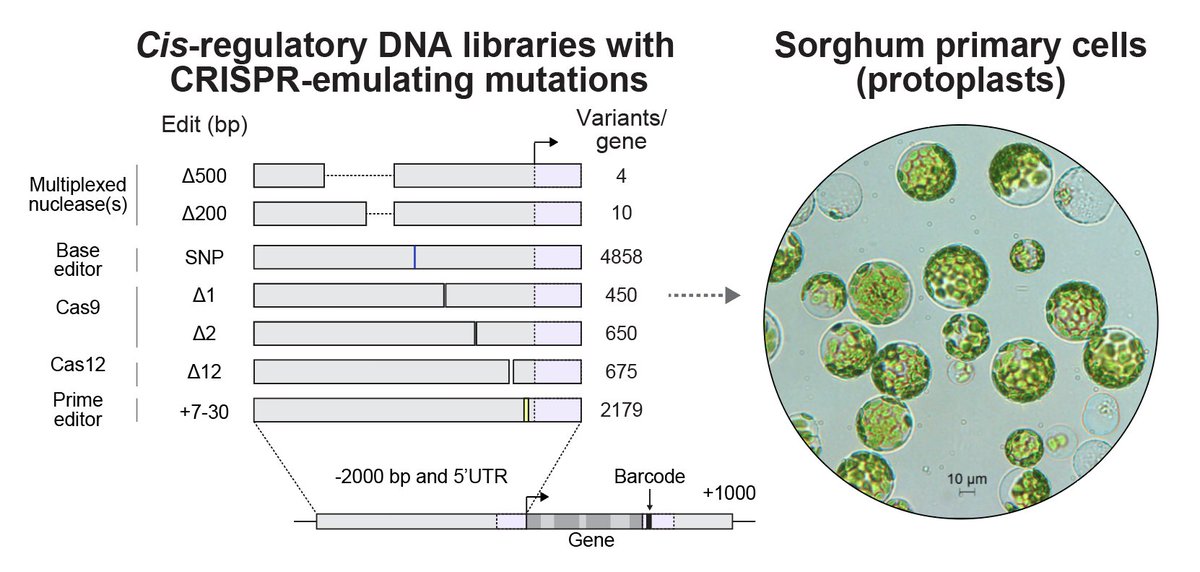
Can CRISPR edits enable precise tuning of plant gene expression? We think: yes.
In our newest manuscript, we measured the effects of >30,000 CRISPR-like promoter mutations in sorghum protoplasts.
protein language models capture rich structural signals, but where that knowledge lives in the network is still unclear
we show that small subnetworks inside PLMs encode structural concepts, from residues to folds
https://t.co/hwG8gaKIo3 @PLOSCompBiol
work led by @riavinod_!
Compressing the collective knowledge of ESM into a single protein language model @naturemethods
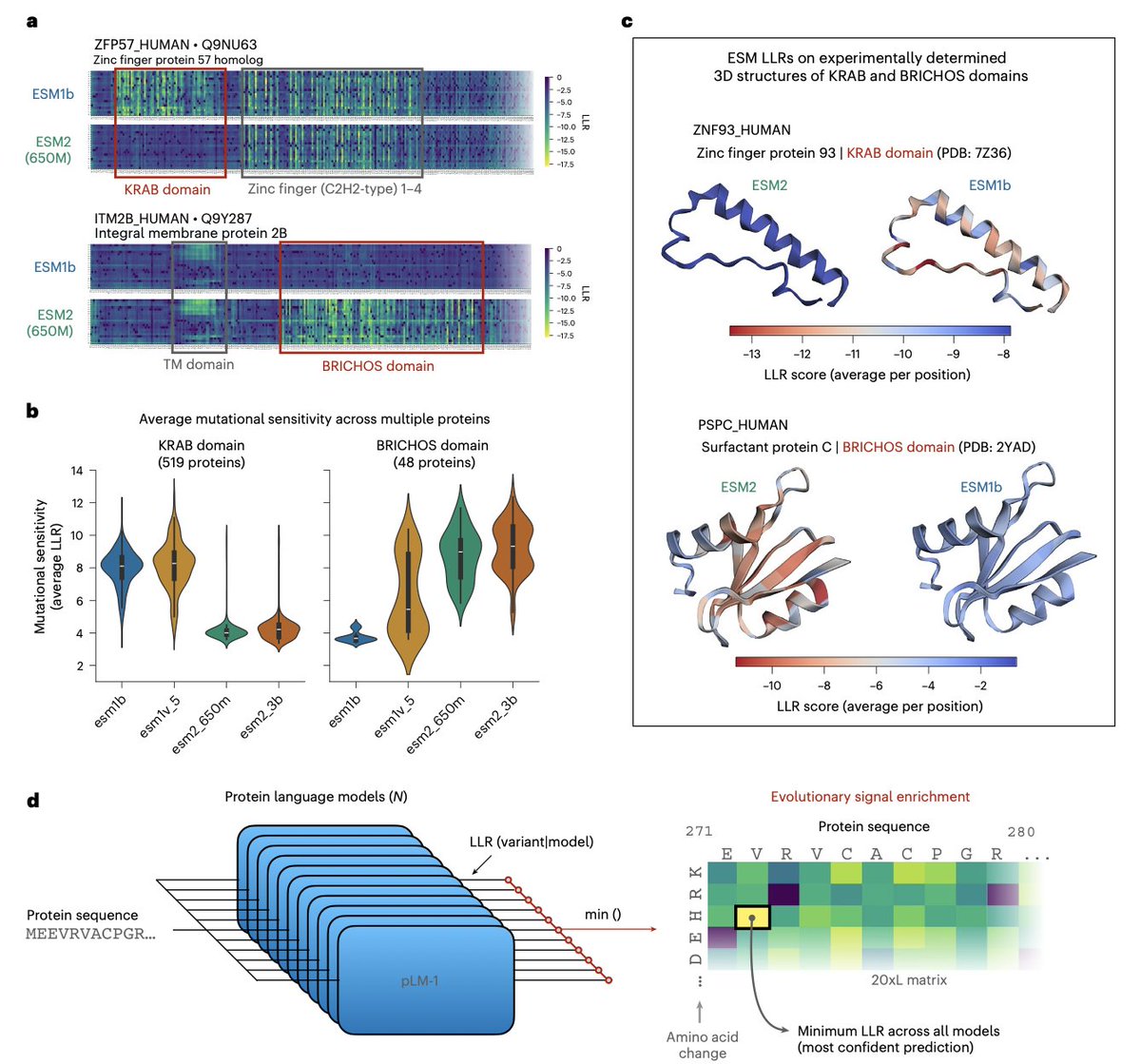
1. The paper argues that “sequence-only” protein language models (PLMs) are not intrinsically capped for variant-effect prediction (VEP); instead, their evolutionary signals are fragmented across model families and can be recovered by making models learn from each other.
2. Key observation: closely related ESM models have complementary blind spots. For example, ESM2 models systematically miss KRAB-domain conservation signals, while ESM1b/ESM1v can miss BRICHOS-domain signals; yet at least one model in the family captures each domain’s mutational sensitivity.
3. They introduce a simple but effective ensemble rule: for each missense mutation, take the minimum log-likelihood ratio (LLR) across models (ESMIN), i.e., “maximum confidence” scoring. This can amplify subtle evolutionary constraints that averaging would dilute.
4. A theoretical analysis explains when min-LLR beats averaging: if pathogenic-variant LLRs are more dispersed across models than benign-variant LLRs (variance asymmetry). The ESM family empirically shows this property, making maximum-confidence aggregation advantageous.
5. ESMIN is evaluated using 11 sequence-only ESM models (ESM1b, five ESM1v, five ESM2; excluding ESM2-15B). It outperforms averaging-based ensembles and improves ProteinGym DMS correlations, with gains occurring in ~50% of assays (versus ~20% for typical ensembles).
6. Main methodological contribution: “maximum-confidence co-distillation.” For each protein, all models score all mutations; the element-wise minimum LLR matrix becomes a teacher signal, and each model is trained (variant-level MSE) to match these confident targets—without MSAs, structures, or population genetics features.
7. Co-distillation substantially improves every participating model, including small ones: ESM2-8M improves on ClinVar AUC from ~0.65 to ~0.88. Several co-distilled single models (e.g., ESM2-3B, ESM1b, ESM2-650M) can even surpass the ESMIN teacher signal (“student surpasses teacher”).
8. Robustness/ablation: improvements persist when training data are heavily reduced and de-homologized. With only ~1% of human proteins (~200 sequences; <30% identity to benchmark proteins), ESM2-35M reaches ~97% (ClinVar) and ~94% (DMS) of its peak co-distilled performance.
9. Iterative procedure: after round 1 (min-LLR co-distillation), additional rounds switch to average-aggregation co-distillation. As models improve, class-conditional variances become more symmetric, making averaging slightly better; after 3 rounds, a single 3B model matches the ensemble—named VESM-3B.
10. Practical compression: VESM-3B is distilled into smaller models (650M, 150M, 35M) that retain most performance (reported as >98% on Balanced ClinVar and >93% on ProteinGym DMS relative to VESM-3B), enabling high-throughput VEP under limited compute.
11. Clinical benchmark (ProteinGym ClinVar, 2,227 genes): sequence-only VESM models outperform other sequence-only PLMs (including ESM-C) and compete with or surpass methods using MSA/structure/population priors. VESM-3B shows balanced ROC behavior across specificity and sensitivity regimes.
12. AlphaMissense comparison: VESM-3B performance is stable across allele-frequency strata, while AlphaMissense shows strong dependence on MAF (consistent with circularity risks when population frequency informs clinical labels). After excluding variants overlapping AlphaMissense training (gnomAD v2 MAF > 1e-5), all VESM sizes outperform AlphaMissense on AUC and multiple calibrated metrics.
13. Modular use of structure: rather than retraining a joint model, they fine-tune the sequence component of ESM3 using VESM-style sequence-based loss to create VESM3, and combine VESM3 with VESM-3B into a structure-aware ensemble (VESM++). This improves performance on structure-dependent DMS assays (binding/stability/expression) while maintaining strong fitness/activity performance.
14. Cross-domain generalization: despite co-distillation being trained on human proteins, gains transfer strongly to nonhuman DMS assays, with disproportionately large improvements reported for viral proteins—even though ESM3’s released training data excluded viral sequences.
15. Beyond binary pathogenicity: using UK Biobank/Genebass summary statistics for 332 gene–phenotype pairs (blood biochemistry biomarkers), variant-level VESM scores correlate with single-variant effect sizes (β). VESM++ and VESM-3B yield the strongest gene–trait association signals across tested models.
16. Notably, VESM-3B recovers the correct pLoF direction of effect in 98.8% of significant gene–phenotype pairs and identifies many associations not detected by missense burden tests, suggesting utility for quantitative trait interpretation from summary statistics.
📜Paper: https://t.co/5gZBiAIx2u
#ProteinLanguageModels #VariantEffectPrediction #ComputationalBiology #HumanGenetics #ESM #ClinVar #ProteinGym #DeepMutationalScanning #UKBiobank #MachineLearning
























![stanleyqilab's tweet photo. "Given a fair wind [epigenetic editing] seems likely to establish itself as an important part of the medicine of the mid-21st century."
@TheEconomist recently ran a feature on epigenome editing, and the framing represents a major shift in mainstream understanding. They positioned the field not as a mere alternative to gene editing, but as the lower-risk, more nuanced path forward for many therapeutic applications.
Reflecting on this as someone who has been in the field since 2012, a few things stand out:
1. The "cutting paradigm" dominated the CRISPR conversation for years. The realization that epigenetic editing’s safer profile is a substantive scientific advantage is finally reaching a broader audience.
2. With three companies in human trials, including the progress we are making at Epicrispr Biotechnologies on FSHD, this is no longer speculative. We are in the era of patient outcomes.
3. The article rightfully flags longevity as a highly speculative frontier. While the epigenetic component of aging is real, we must remain ambitious without overpromising as we move from treating disease to enhancing human healthspan.
To the founders, clinicians, and especially the students and postdocs: this recognition is a signal that the work of the past decade has built something real.
The next decade will be even more transformative.
The article: https://t.co/ZSbv2Af53I
#EpigeneticEditing #Biotech #CRISPR #GeneTherapy #FSHD #EpicrisprBiotechnologies #Stanford](https://pbs.twimg.com/media/HH0EHTHa0AA6IFi.jpg)